META-Insight
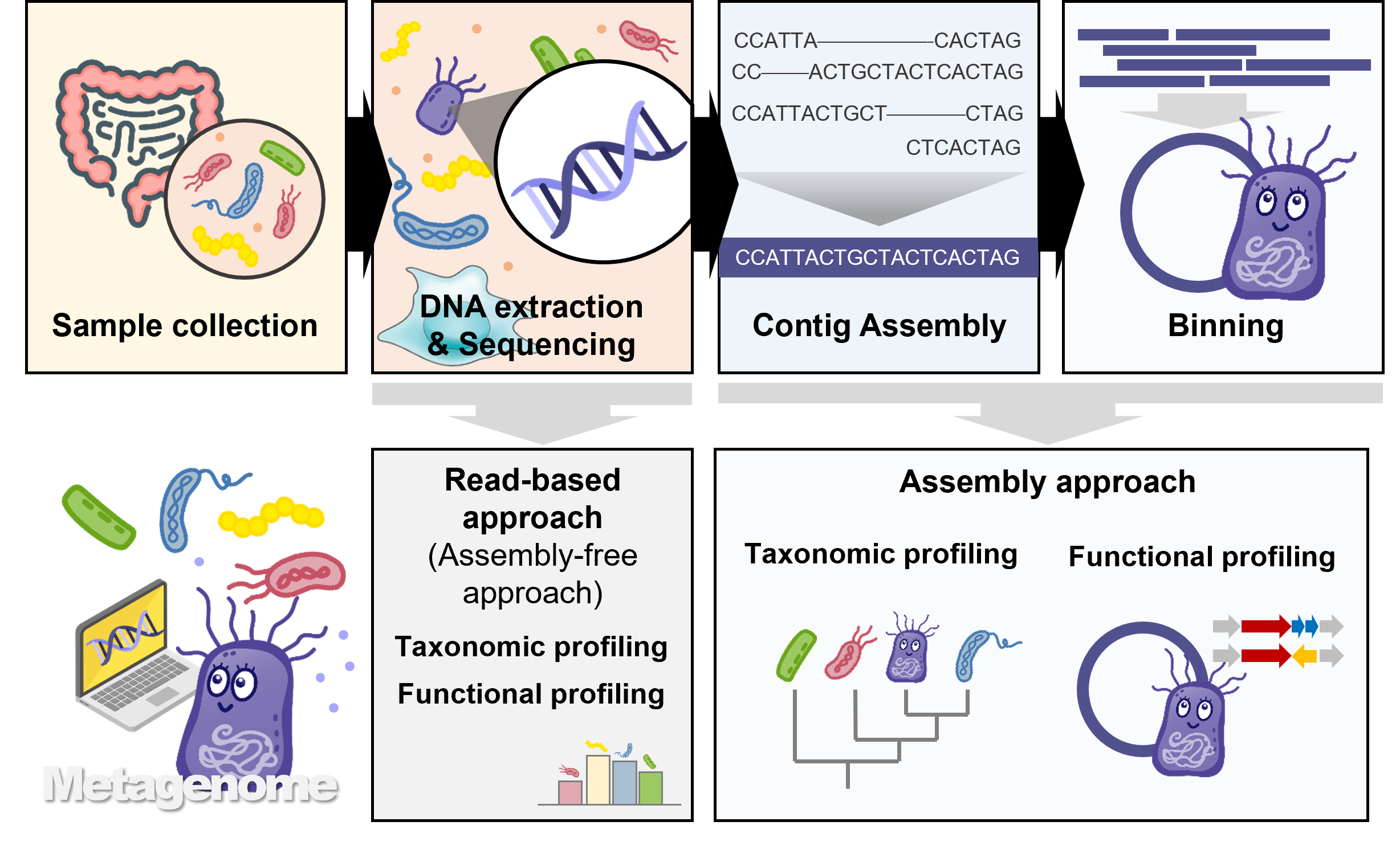
Metagenomic analysis is an innovative approach to interpreting complex biological information to understand the ecology, function, diversity, and interactions of microbial communities.
Our goal is to introduce state-of-the-art analytical methods that enable accurate meta-omics analysis and provide meta-omics data derived from the accurate gut microbiome of healthy individuals.
The key feature of our data analysis method is that it organizes microbial community information at the level of microbial genomes, mainly focusing on metagenome-assembled genomes (MAGs).
Additionally, while metagenomes can offer potential functional information, the presence of specific genes does not necessarily imply their expression.
By combining metatranscriptome and metaproteome data, we can confirm gene expression or suppression under specific conditions, allowing us to distinguish active microbes from inactive ones from a metabolic perspective.
In particular, at each step of NGS meta-omics analysis, we introduce an analysis based on shotgun sequencing for metagenome, RNA-seq for metatranscriptome, and Ribo-seq data for metaproteome. In addition to meta-omics data analysis protocols, Meta-Insight provides precise meta-omics data of the gut microbiome in healthy individuals.
Such data can be helpful not only in understanding the functions of the gut microbiome but also in serving as a control group for research on gut microbiomes related to diseases.
PROTOCOLS
Meta-Insight provides a metagenome analysis protocol that focuses on MAG composition. When performing de novo genome analysis through shotgun sequencing, a process of assembling short reads is required.
Similar to assembling a single genome from these short fragments, information assembled for each microbial unit in a complex microbial community can offer much more insight into the microbiome ecology.
Here, we introduce both Illumina's short read and PacBio’s long read-based analysis methods.
In terms of metatranscriptomics, our analysis protocol includes both MAG-based reference-guided analysis and reference-independent analysis. We describe a method to profile specific expression levels of genes, MAGs, and also detail DE analysis to find differentially expressed genes for experimental design to explore expression level differences.
One alternative method designed for mass-based analysis of metaproteomic data is Ribo-seq.
This method involves fixing the cell state by treating it with chloramphenicol, removing unprotected nucleic acids through MNase treatment, and then analyzing the mRNA sequences involved in protein synthesis. We expect that this approach can address issues associated with low throughput and limited accuracy in databases in bulk analysis-based methods.
Additionally, protein synthesis can be quantified by identifying the genetic information coding for the protein.
The tool selected in the metaomics analysis protocol produces reproducible output, and in cases with similar specifications, software was selected for general use and good computing efficiency.
In the case of the database used, we selected one that was well curated and capable of regular updates.
GENOME/GENE/SAMPLE CATALOG

GENOME CATALOG: Provides information on the healthy individual's gut microbiome at the core metagenome-assembled genome (MAG) level.
GENE CATALOG: Provides information on the healthy individual's gut microbiome at the gene level, including the normalized abundance of each gene and the expression levels of transcripts and proteins.
BLAST: Allows user access based on sequence (nucleotide/amino acid).
SAMPLES: Provides information about the cohort used for database construction, including metadata and network of key microbial groups.


